何兵 练宇翔 吴木生† 罗文崴 杨慎博 欧阳楚英‡
1) (江西师范大学物理与通信电子学院,南昌 330022)
2) (鸿之微科技(上海)股份有限公司,上海 200120)
三元锂金属卤化物作为极具潜力的固体电解质材料受到人们的广泛关注.本文利用基于密度泛函理论的第一性原理方法研究了一系列具有不同Li 离子浓度的LixYCl3+x (x=2.14,3.00,4.20)和LixYBr3+x (x=1.8,3.0,5.0)材料的结构、电子性质和迁移特性.研究结果表明,Li 离子和Li 空位浓度的变化对材料的性能有显著影响,而且随着x 值的增加,Li 离子的含量增加,相应的Li 空位浓度降低,结构的稳定性增强、带隙增大、离子迁移能垒降低,从而可以调控该类材料的性能.此外,计算结果进一步表明,在所有考虑的结构中,具有最佳Li 离子与Li 空位平衡的Li3YCl6 和Li3YBr6 组分展现出最高的结构稳定性、最大的带隙和最低的迁移能垒.本文为设计性能更好的卤化物固态电解质提供了一种新策略和新思路.
与传统商业锂离子电池相比,包含无机固态电解质的全固态锂离子电池由于具有更高的安全性、更大的能量密度、更好的稳定性被认为是动力电池和下一代能源存储设备的首选,受到人们的广泛关注[1,2].研发具有高能量密度的全固态锂离子电池,要求开发具有良好化学和电化学稳定性的高Li 离子导电的固态电解质.然而,开发室温下可与液体电解质相媲美、兼顾高锂离子电导率和高稳定性的固态电解质,一直是全固态锂离子电池发展面临的一个巨大挑战.当前,虽然一批体相室温离子电导率超过10—3S/cm,甚至达到10—2S/cm 的氧基或硫基的无机固态电解质材料相继问世[3-5],但遗憾的是,这些电解质材料在离子电导率和化学稳定性之间很难平衡,从而阻碍了其在全固态锂离子电池中的应用.例如,对于典型的快离子导体材料—Li10GeP2S12,研究发现其电化学窗口仅为0.57 V(1.72—2.29 V),无法与目前的电极材料兼容;而电化学窗口相对较大的氧化物材料不仅电导率普遍比硫化物低1 个数量级,而且与正极材料之间存在较大的接触电阻,导致全电池的倍率性能较差[6].此外,通过高通量筛查进一步发现,氧、硫化物电解质材料与电极的界面问题是普遍现象[7].由此可见,寻找同时满足高离子电导率和高稳定性的固态电解质材料仍然是目前全固态锂电池发展的关键方向之一.
针对上述电解质面临的挑战,人们重新将目光转移到具有本征氧化稳定性和机械形变性的卤化物材料上[8].首先,一价卤素阴离子理论上与Li 离子的相互作用要比二价S 或O 离子的弱,这将减弱Li 离子与阴离子配体间的库仑相互作用,从而有望实现Li 离子的快速迁移.其次,六配位卤素阴离子的离子半径(rCl-=1.67 Å,rBr-=1.82 Å,rI-=2.06 Å,1 Å=10—10m)要明显大于O2—离子(rO2-=1.26Å)半径[9],原则上有利于扩大Li 离子的迁移通道[10].基于卤素的这些本质特征,实际上早在几十年前,Lutz等[11-13]对各种结构和组分的氯化物(如Li2MgCl4,Li2ZnCl4,Li3YCl6,Li3InCl6等)固体电解质进行了广泛研究,但是由于这些早期卤化物展现出比较低的室温电导率(约10—6S/cm),因此对卤化物电解质材料的研究进展非常缓慢.然而,随着制备工艺及合成方法的不断改进,近几年,一类具有Li3MX6(M代表Sc,Y,In 或稀土金属;X代表卤素)通式的三元卤化物的研究取得了重大进展,越来越多满足要求的三元卤化物快离子导体材料被成功合成.例如,Asano等[14]采用机械研磨法制备的Li3YCl6和Li3YBr6材料的室温离子电导率均达到10—3S/cm 数量级,同时它们和高电压(> 4 V)正极间表现出良好的氧化稳定性.随后,西安大略大学孙学良课题组利用机械研磨[15]和水相合成[16]方法都重新合成了高性能的Li3InCl6,该材料除了呈现出2×10—3S/cm 的高室温离子电导率,还展现出对空气/湿度的稳定性.这些新兴的三元卤化物,由于在成本、稳定性和质量生产率方面要优于硫化物材料,在离子导电性方面要优于氧化物,因而引起了人们极大的兴趣.更有趣的是,通过理论研究进一步发现,这类由密堆积的卤素阴离子通过离子键结合的三元卤化物,展现出来的高离子电导率和高氧化稳定性是其固有属性,即卤素离子类型及空间排布对其电导率的影响较小[17].这与只有体心立方(body-centered cubic,BCC)阴离子框架才能呈现高离子电导率和低活化能的氧、硫化物相比[18],将极大地降低材料的设计和制备难度.最近,一系列具有高离子电导率(约10—3S/cm)和氧化稳定性的、不同卤素离子和排布的Li3MX6(LMX)材料,通过不同的合成方法成功制备,这进一步表明合成条件和晶体结构细节可能对这类材料的离子传导发挥了重要作用[19,20].
然而,到目前为止,对于这类卤化物的最佳合成细节,特别是最佳的Li 离子和Li 空位浓度仍不十分清楚,这将阻碍其性能的改善及新材料的设计.如通过传统高温合成的早期Li3YCl6样品的Li 离子室温电导率约为10—5S/cm[13],比用新工艺合成的相同材料[14]展现出的电导率(约10—3S/cm)低得多.这种由于不同合成方法和条件产生的性能差异,表明在合成过程中调整迁移率的能力,成为进一步研究该材料的必要条件.2020 年,孙学良课题组[21]通过调控ScCl3和LiCl 的不同摩尔比,成功制备了一系列具有C2/m空间群、不同Li 离子和Li 空位浓度的LixScCl3+x(x=2.5,3.0,3.5,4.0)材料,并获得了具有3×10—3S/cm 室温电导率的最佳组分Li3ScCl6,表明调控反应物的含量可以改善LixScCl3+x材料的导电性,且存在最佳的Li 离子和Li 空位浓度.然而,这一发现是否适合其他LMX材料,特别是被广泛研究的Li3YCl6和Li3YBr6材料,目前仍不清楚.虽然最近一系列研究发现,通过对Li3YCl6材料进行Li/Y 占位调控、Y 位掺杂或卤素混合来调控Li 离子浓度或晶体结构,可以进一步改善材料的离子电导率[22-24],但是目前仍不清楚Li 离子和Li 空位浓度对这种有着相同阴离子框架、不同Li/M阳离子构型的材料究竟具有哪些影响? 另外,北京大学孙强和马里兰大学莫一非课题组[17]共同研究发现,对于相同阳离子配置和浓度的Li3YCl6,第一性原理分子动力学(ab initiomolecular dynamics,AIMD)预测的室温离子电导率(1.4×10—2S/cm)比实验值(0.5×10—3S/cm)[14]高了近2 个数量级;而Li3YBr6的理论模拟结果(2.2×10—3S/cm)[17]却与实验值(1.7×10—3S/cm)[14]相当符合.这种差异是否与制备过程中的Li 离子和Li 空位浓度的调控有关,仍需进一步证实.由此可见,尽管Li3YCl6和Li3YBr6被广泛关注,但二者调控电导率的关键因素仍需深入研究,而Li/M阳离子浓度和构型对离子导电的影响是需要考虑的因素之一.
第一性原理计算已经被广泛证实,能在原子层面上较好地理解Li 离子扩散机理以及设计和预测新型锂离子电池材料[25-27].本文利用第一性原理计算系统研究了具有不同Li 离子和Li 空位浓度的LixYCl3+x(x=2.14,3.00,4.20)和LixYBr3+x(x=1.8,3.0,5.0)材料中,Li 离子和Li 空位浓度对其性能的影响,以了解这类系统中Li 离子浓度对其快速扩散的决定程度,并预测最佳的Li 离子和Li 空位浓度,为优化实验合成条件提供参考.结果表明,阳离子构型和Li 离子浓度显著影响这类材料的离子电导率,且在所有组分中,Li 离子和Li 空位存在最佳平衡浓度,而Li3YCl6和Li3YBr6两种配置均具有最佳的性能,进一步证实了实验的最佳合成组分.鉴于卤化物阴离子化学之间的相似性,本文为设计新型LMX 类卤化物,并为改善电导性提供了指导.
本文所有计算均采用基于密度泛函理论(density functional theory,DFT)的第一性原理计算方法,并在VASP (Viennaab initiosimulation package)[28]和DS-PAW[29]软件包中实现.通过缀加投影平面波赝势(projector-augmented wave method,PAW)[29]描述原子实和价电子之间的相互作用,展开平面波的截断能设置为550 eV,电子间相互作用采用广义梯度近似(generalized gradient approximation,GGA)的Perdew-Burke-Ernzerh(PBE)交换交联泛函描述[30].考虑到过渡金属元素Y 的d 电子间的强关联效应,我们采用Dudarev等[31]提出的Hubbard 修正(GGA+U)的方法在DS-PAW 软件中进行结构弛豫和静态计算,且有效U值(Ueff)取为3.00 eV[32].弛豫时布里渊区的数值积分采用Monkhorst-Pack 方法[33],所有结构的k点采样密度约为0.028/Å—3.Li,Y,Cl 和Br 原子的价电子配置分别为2s12p0,4s24p64d15s2,3s23p5和4s24p5.晶胞内所有原子进行完全弛豫,弛豫收敛精度为1×10—5eV/atom,原子间相互作用力的收敛精度为1×10—2eV/Å.
Li 离子低能迁移路径的搜索及相应迁移能垒的计算采用基于过渡态理论的弹性能带(nudge elastic band,NEB)方法[34].NEB 方法在锂离子电池材料中,被证明是寻找离子扩散能量鞍点的一种有效方法[35-37].对于Li2.14YCl5.14和Li4.2YCl7.2组分的基态结构,采用CALYPSO (crystal structure analysis by particle swarm optimization)软件[38,39]进行结构搜索,以获得低能基态构型.本工作的所有计算流程由电池材料高通量计算平台管理[25].
3.1 晶体结构
为了准确研究Li 离子浓度对材料导电性的影响,首先需获得所有组分的基态结构.基于实验结果,Li3YCl6和Li3YBr6分别是具有和C2/m空间群的三角和单斜结构[14],且Li3YCl6和Li3YBr6中的阴离子框架分别为六角密堆积(hexagonal closed-packed,HCP)和立方密堆积(cubic closedpacked,CCP).Li3YCl6中的Cl 原子满占据6i位,Li 原子部分占据在6g和6h位,Y 原子满占据1a位、部分占据2d位;而Li3YBr6中的Br 原子满占据8j和4i位,Li 原子满占据4g位、部分占据4h位,Y 原子部分占据2a和2d位.为了能更清晰地构建各组分的模型,我们先建立了Li,Y 和Cl/Br 原子都满占据对应Wyckoff 位的单胞结构模型,即有着Li12Y5Cl18和Li8Y4Br12分子式的结构单元,分别如图1(a)和图2(a)所示.
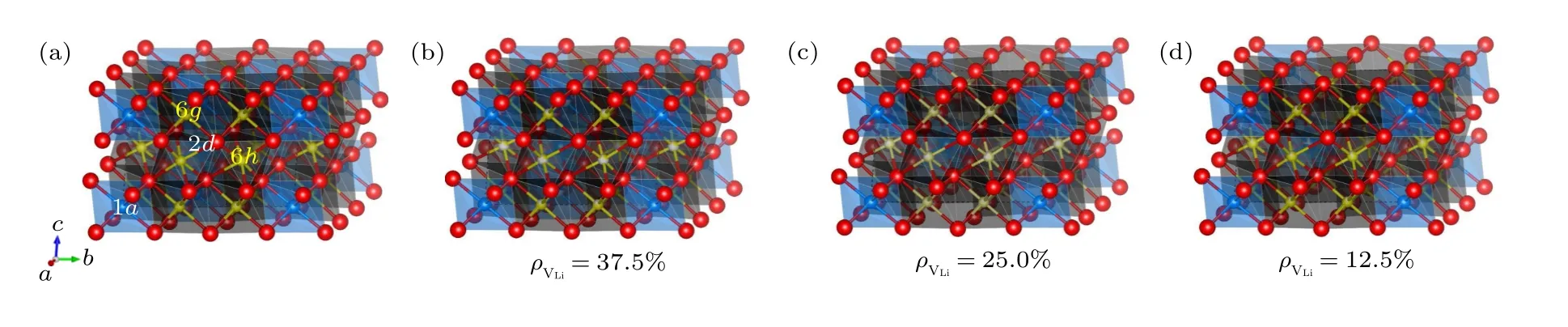
图1 原子结构示意图 (a) Li12Y5Cl18;(b) Li7.5Y3.5Cl18;(c) Li9Y3Cl18;(d) Li10.5Y2.5Cl18 (黄色、蓝色和红色圆球分别代表Li,Y 和Cl 原子,有部分白色的小球代表对应原子在该位置的部分占据)Fig.1.Schematic diagram of atomic structures: (a) Li12Y5Cl18;(b) Li7.5Y3.5Cl18;(c) Li9Y3Cl18;(d) Li10.5Y2.5Cl18 (The yellow,blue and red spheres represent Li,Y and Cl atoms,the spheres with a partially white indicate the partial occupancy of the corresponding atoms).
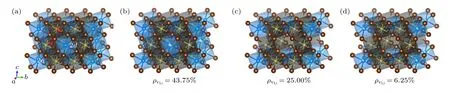
图2 原子结构示意图 (a) Li8Y4Br12;(b) Li4.5Y2.5Br12;(c) Li6Y2Br12;(d) Li7.5Y1.5Br12 (黄色、蓝色和咖啡色圆球分别代表Li,Y 和Br 原子,有部分白色的小球代表对应原子在该位置的部分占据)Fig.2.Schematic diagram of atomic structures: (a) Li8Y4Br12;(b) Li4.5Y2.5Br12;(c) Li6Y2Br12;(d) Li7.5Y1.5Br12 (The yellow,blue and brown spheres represent Li,Y and Br atoms,the spheres with a partially white indicate the partial occupancy of the corresponding atoms).
为简便起见,在Device Studio 软件平台上[40]以Li12Y5Cl18和Li8Y4Br12结构为基础,按照实验中Y 原子和Li 原子的占位权重[14],通过删减不同数量的Y 和Li 原子来构建Li 空位浓度()分别为37.5%,25%和12.5%的LixYCl3+x(x=2.14,3.00,4.20)结构,以及ρVLi分别为43.75%,25%和6.25%的LixYBr3+x(x=1.8,3.0,5.0)结构.以Li9Y3Cl18(Li3YCl6)为例,为了满足化学计量比,Li9Y3Cl18需要在Li12Y5Cl18结构中分别删除3 个Li 原子和2 个Y 原子,由于实验中Li3YCl6高晶样品Y 占据(001)平面内2d位的权重最低,仅为12%左右[14],所以我们在此配置中仅考虑了删除该位上的2 个Y 原子;对于Li 原子,由于6g位的权重高于6h位,所以考虑了同时删除2 个6h位和1 个6g位的Li 原子,其可能的配置构型如图1(c)所示,Y 原子除占据1a位外,还满占据(002)平面内的2d位,Li 离子在6g位的占据权重为83.33%,6h位则为66.67%.相比之下,Li7.5Y3.5Cl18结构中的Y 原子还以50%的权重占据(001)平面内的2d位;而Li10.5Y2.5Cl18的Y 原子则以50%的权重占据(002)平面内的2d位,如图1(b),(d)所示.为了构建上述模型的结构,我们均采用1×1×2 的Li7.5Y3.5Cl18,Li9Y3Cl18和Li10.5Y2.5Cl18超胞来构造所有的配置.采用Li12Y5Cl18类似的处理方法,构建了对应Li4.5Y2.5Br12,Li6Y2Br12和Li7.5Y1.5Br12单胞的LixYBr3+x(x=1.8,3.0,5.0)结构,如图2(b)—(d)所示,并采用2×1×1 的Li4.5Y2.5Br12,Li6Y2Br12和Li7.5Y1.5Br12超胞来构造所有的配置.
正如前面所提及的LixYCl3+x(x=2.14,3.00,4.20)和LixYBr3+x(x=1.8,3.0,5.0)均存在Li或Y 阳离子的部分占据,所以这些结构存在多种构型.要获得基态结构,需要比较所有构型的总能.图3 显示了LixYCl3+x(x=2.14,3.00,4.20)和LixYBr3+x(x=1.8,3.0,5.0)的超胞中所有可能构型的总能.由于Li15Y7Cl36和Li21Y5Cl36的组分中不仅存在6g和6h位Li 的随机占据,而且2d位的Y 也存在随机占据,所以涉及的构型数量非常多.对此,采用CALYPSO 工具来搜索这两种组分的低能结构,所有构型的能量如图3(a),(c)所示.对于Li18Y6Cl36的配置,由于Y 原子的占位确定,所以根据对称性,选择了15 种构型,并直接进行DFT 的总能计算,结果见图3(b).而所有组分的LixYBr3+x(x=1.8,3.0,5.0)结构,由于Li 均是满占据4g位[14],其可能的配置仅有10 多种构型,由此也直接进行了DFT 计算,结果如图3(d)—(f)所示.然后,我们选择所有组分中的最低能构型作为其基态结构,并且利用DS-PAW软件对所有最低能的构型进行了DFT 的完全弛豫,弛豫后的几何结构如图4(a)—(c)和图5(a)—(c)所示,其对应的晶格参数列在表1 中.由表1 可知,本文计算的Li3YCl6和Li3YBr6的晶格常数与实验结果[14]一致.此外,根据LixYCl3+x(x=2.14,3.00,4.20)以及LixYBr3+x(x=1.8,3.0,5.0)的基态结构可得出,Y 和Li 的占位不尽相同,而且随着x的增加Li 离子浓度增加,Li 空位浓度减少,Y 的浓度也随之降低,后续我们将研究这种变化对其性能的影响.
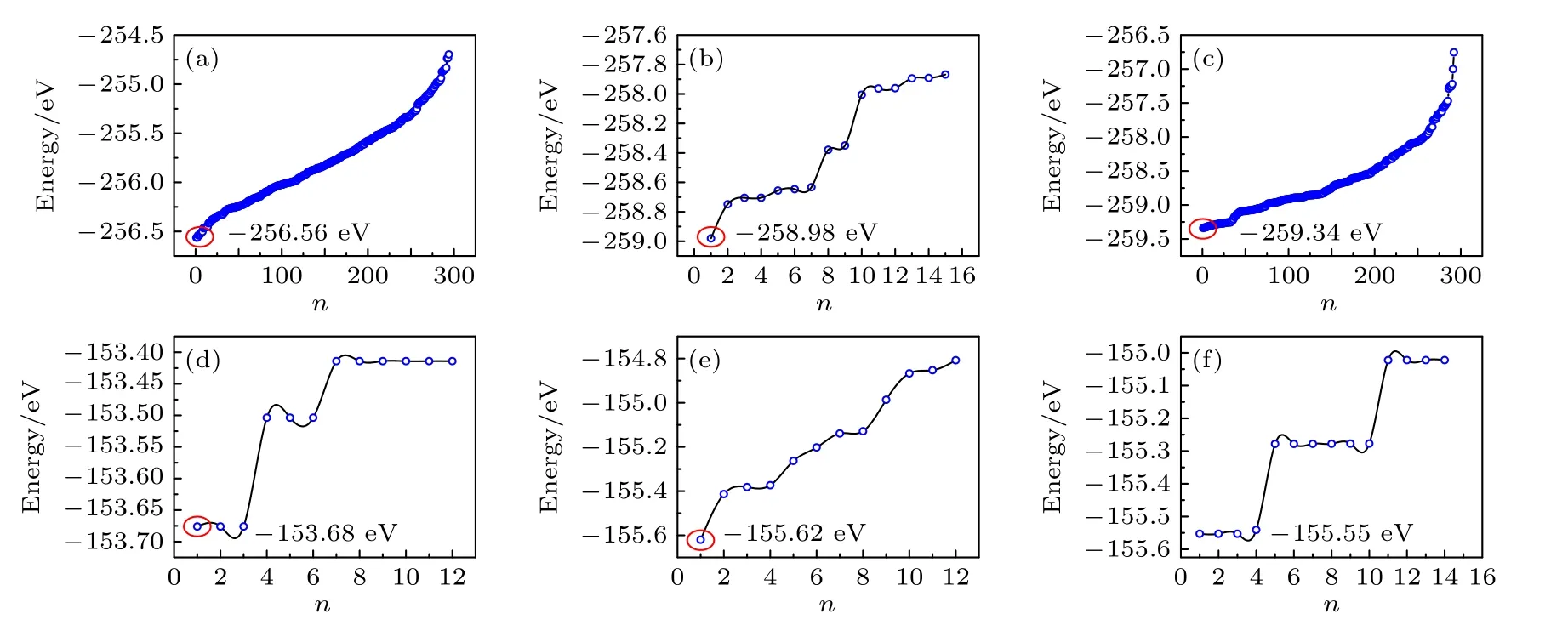
图3 超胞中所有可能构型n 的总能.1×1×2 超胞 (a) Li7.5Y3.5Cl18;(b) Li9Y3Cl18;(c) Li10.5Y2.5Cl18.2×1×1 超胞 (d) Li4.5Y2.5Br12;(e) Li6Y2Br12;(f) Li7.5Y1.5Br12Fig.3.The total energies of all possible configurations n.1×1×2 supercell: (a) Li7.5Y3.5Cl18;(b) Li9Y3Cl18;(c) Li10.5Y2.5Cl18.2×1×1 supercell: (d) Li4.5Y2.5Br12;(e) Li6Y2Br12;(f) Li7.5Y1.5Br12.
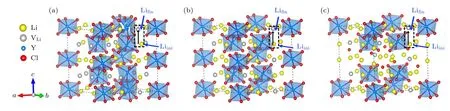
图4 DFT 计算的不同构型的基态结构 (灰色小球代表Li 空位,黑色箭头代表虚线框中的Li 离子向Li 空位迁移的迁移路径)(a) Li15Y7Cl36;(b) Li18Y6Cl36;(c) Li21Y5Cl36 Fig.4.Ground-state structures of different configurations calculated by DFT (The grey spheres represent Li vacancies,black arrows represent the migration paths of Li ions in the dashed box to Li vacancies): (a) Li15Y7Cl36;(b) Li18Y6Cl36;(c) Li21Y5Cl36.
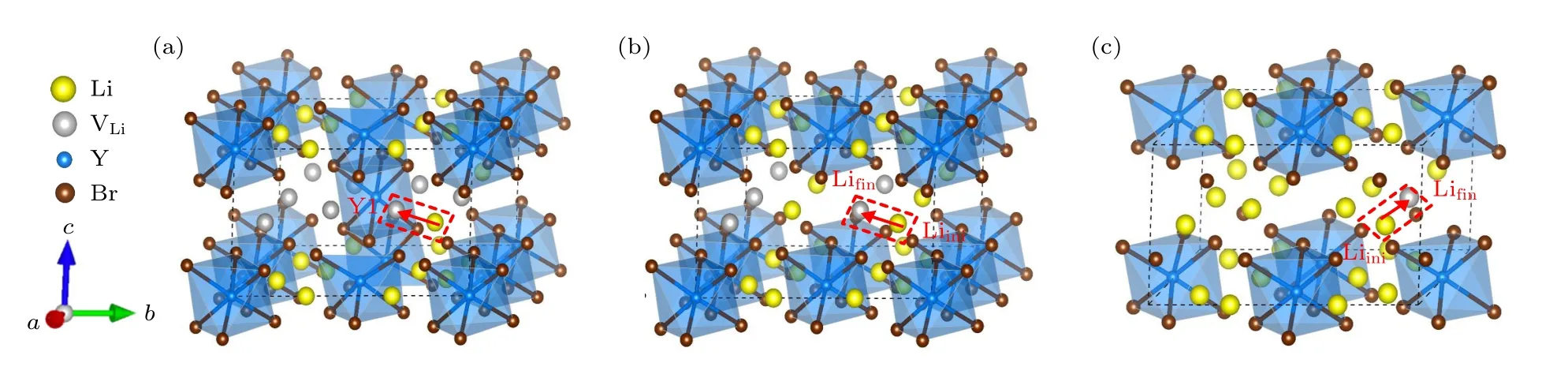
图5 DFT 计算的不同构型的基态结构 (红色箭头代表虚线框中的Li 离子向Li 空位迁移的迁移路径) (a) Li9Y5Br24;(b) Li12Y4Br24;(c) Li15Y3Br24Fig.5.Ground-state structures of different configurations calculated by DFT (Red arrows represent the migration paths of Li ions in the dashed box to Li vacancies): (a) Li9Y5Br24;(b) Li12Y4Br24;(c) Li15Y3Br24.
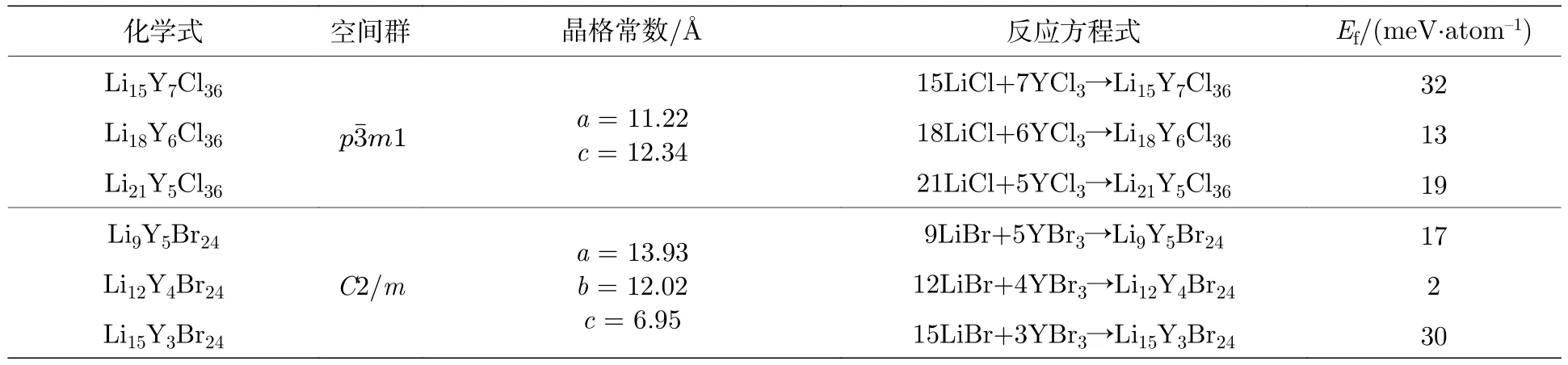
表1 LixYCl3+x 和LixYBr3+x 卤化物的形成能Table 1.Formation energies of LixYCl3+x and LixYBr3+x halides.
在获得所有组分的基态结构后,基于实验合成过程,我们通过如下反应方程,利用DFT 计算来预测所有组分在0 K 下的稳定性:
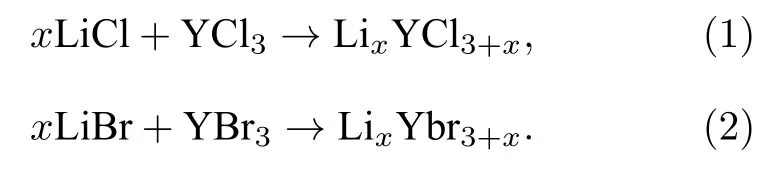
并计算所有组分的形成能Ef,表达式为
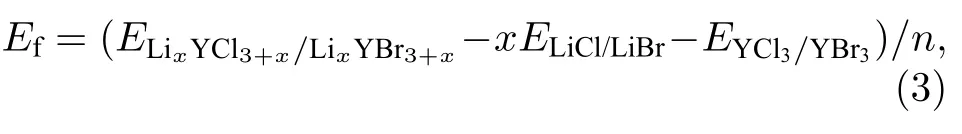
3.2 电子结构
众所周知,作为全固态锂离子电池的电解质材料,宽电化学窗口是需要满足的必要条件之一,而理论电化学窗口的上限主要取决于材料的带隙.为了估算所有组分的理论电化学窗口,我们利用GGA方法计算了所有组分的的电子态密度(denstiy of state,DOS),如图6 所示.从DOS 图可以看到,6 种材料都是宽带隙的绝缘体,其带隙宽度都在3.58—5.43 eV 内,考虑到一般而言GGA 方法对带隙的低估,其理论电化学窗口应在5 V 以上,表明所有组分都具有宽的理论电化学窗口.进一步观察图6 中的部分态密度(partial density of state,PDOS),可以看到,所有组分的价带顶(valence band maximum,VBM)和导带底(conduction band minimum,CBM)均分别主要由卤素离子(Cl—/Br—)和Y3+离子的电子态构成,这意味着在该电解质材料中优先参与氧化反应的是Cl—/Br—阴离子,优先参与还原反应的是Y3+,考虑到Cl 和Br 卤素原子的强电负性以及Y 和Li 的强还原性,不难理解该类材料具有宽电化学窗口的原因.显然,在LixYCl3+x(x=2.14,3.00,4.20)中,Li18Y6Cl36具有最宽的带隙(5.43 eV),其次是Li21Y5Cl36,带隙最小的是Li15Y7Cl36,见图6(a)—(c),表明在这三种组分中,Li3YCl6具有最宽的理论电化学窗口,即理论上具有最高的电化学稳定性.同理,如图6(d)—(f)所示,对于LixYBr3+x(x=1.8,3.0,5.0)材料,其带隙遵循Li12Y4Br24(4.69 eV) > Li15Y3Br24(4.00 eV) >Li9Y5Br24(3.58 eV)的顺序,Li3YBr6具有最高的电化学稳定性.
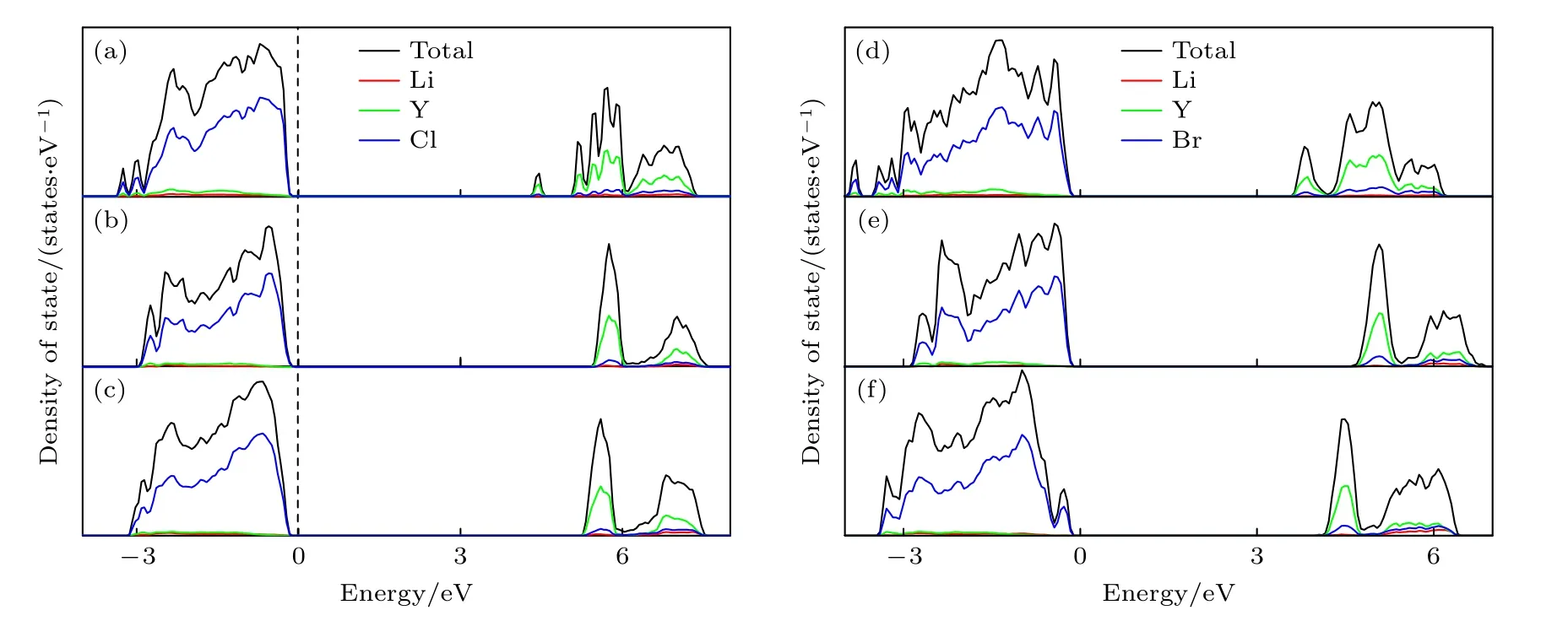
图6 所有组分的DOS 图及带隙 (a) Li15Y7Cl36 (4.35 eV);(b) Li18Y6Cl36 (5.43 eV);(c) Li21Y5Cl36 (5.33 eV);(d) Li9Y5Br24 (3.58 eV);(e) Li12Y4Br24 (4.69 eV);(f) Li15Y3Br24 (4.00 eV)Fig.6.DOS plots and band gap results for all components: (a) Li15Y7Cl36 (4.35 eV);(b) Li18Y6Cl36 (5.43 eV);(c) Li21Y5Cl36(5.33 eV);(d) Li9Y5Br24 (3.58 eV);(e) Li12Y4Br24 (4.69 eV);(f) Li15Y3Br24 (4.00 eV).
3.3 Li 离子迁移特性
在锂离子电池中,Li 离子迁移快慢是反映电池倍率性能的一个关键参数,因此研究Li 离子的迁移能垒对电解质材料的应用至关重要.在固体电解质材料中,Li 离子的电导率通常受到各种各样本质因素的影响,例如,离子特性 (传导和骨架离子半径、电荷、电负性等)、传导机理 (空位迁移、间隙跃迁、协同输运等)、空间排布和配位环境等[44,45].根据前面的基态结构分析可知,在LixYCl3+x(x=2.14,3.00,4.20)或LixYBr3+x(x=1.8,3.0,5.0)离子晶体中,组分间的不同之处主要体现在阳离子占位及浓度,因此影响其Li 离子迁移能垒的因素主要体现在Li 离子与周围离子之间的库仑作用强度.由此,主要从这个角度分析所有组分材料中Li离子的迁移机理.根据我们以前的NEB 计算结果[46],发现 Li3YCl6是具有0.19 eV 迁移能垒的沿c方向的一维扩散通道,而Li3YBr6是具有0.24 eV 各向同性的三维扩散通道.然而,在另外两种组分的同类材料中,即LixYCl3+x(x=2.14,4.20)材料或LixYBr3+x(x=1.8,5.0)材料,Li 离子的扩散路径是否与Li3YCl6或Li3YBr6相同,仍需要进一步研究.
为了进一步证实Li3YCl6和Li3YBr6中Li 离子的扩散通道,我们利用能快速有效地揭示离子晶体中Li 离子扩散通道的键价位能(bond valence site energy,BVSE)方法[47,48]计算了Li18Y6Cl36和Li12Y4Br24中的Li 离子传输通道,分别如图7(a)和图8(a)所示,黄色区域是Li 离子的分布密度,代表Li 离子导电通道,黄色区域包围的浅黄色小球是稳定的Li 位.图7 和图8 所有的晶体结构和等位面图均由VESTA 软件可视化[49],等值面分别为0.8e,0.7eÅ—3.对于Li18Y6Cl36,由图7(a)可知,稳定的Li 位在八面体位,且Li 离子在c方向的分布密度是连续的,而在ab平面内是非连续的,说明其具有沿c方向的一维传导通道(图7(a)中的黑色箭头所示).显然,该通道是由c方向相邻的LiCl6八面体直接连接,如图7(a)中的椭圆区域插图所示,LiCl6形成的Oct2 或Oct1 八面体与近邻的Li 空位(VLi,绿色小球)八面体构成了Li 离子的扩散通道.不同于Li18Y6Cl36,由图8(a)可知,在Li12Y4Br24中,三个方向的黄色区域都是连续的,表明其Li 离子具有三维传导通道,并且三个方向的Li 离子传导路径都是通过LiBr4四面体间隙位相连(Oct1-Tet1-VLi,Oct2-Tet2-VLi,Oct3-Tet3-VLi),如图8(a)中的椭圆形插图所示.这些BVSE计算的结果与我们NEB 的结果[46]一致.同样,我们利用BVSE 分别计算了Li15Y7Cl36和Li21Y5Cl36,Li9Y5Br24和Li15Y3Br24中Li 离子的传导路径,如图7(b),(c)和图8(b),(c)所示.BVSE 的计算结果显示,Li15Y7Cl36和Li21Y5Cl36具有与Li18Y6Cl36相同的Li 离子传导通道,即同样具有沿c方向的一维扩散路径.同样地,Li9Y5Br24和Li15Y3Br24具有与Li12Y4Br24相同的三维传导通道.
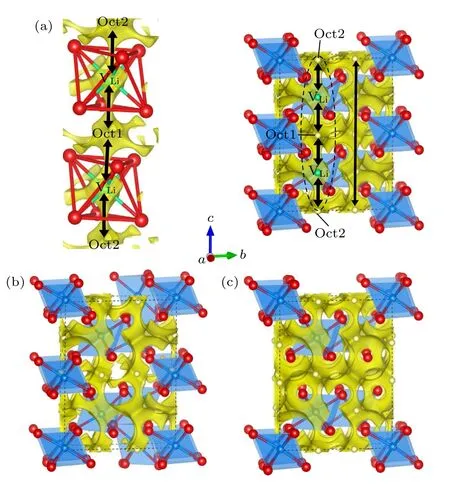
图7 (a) Li18Y6Cl36,(b) Li15Y7Cl36,(c) Li21Y5Cl36 的晶体结构与BVSE 计算的Li 离子等位面势图的叠加图(黄色区域是Li 离子的分布密度,代表Li 离子的传导通道)Fig.7.Crystal structures of (a) Li18Y6Cl36,(b) Li15Y7Cl36,(c) Li21Y5Cl36 superimposed with the Li-ion potential map calculated using BVSE (The yellow isosurfaces correspond to Li-ions probability density,indicating Li-ion conduction path).
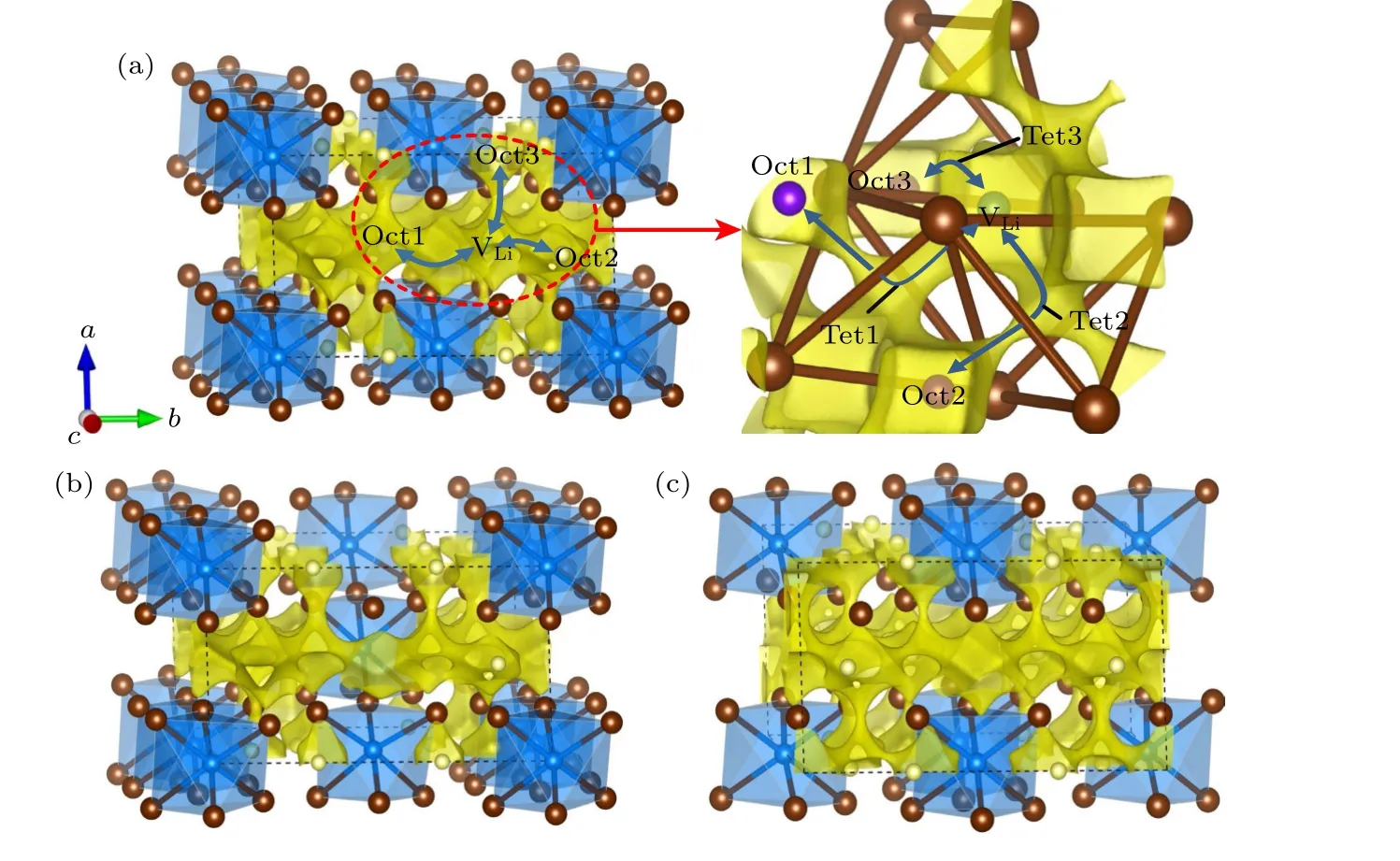
图8 (a) Li12Y4Br24,(b) Li9Y5Br24,(c) Li15Y3Br24 的晶体结构与BVSE 计算的Li 离子等位面势图的叠加图(黄色区域是Li 离子的分布密度,代表Li 离子的传导通道)Fig.8.Crystal structures of (a) Li12Y4Br24,(b) Li9Y5Br24,(c) Li15Y3Br24 superimposed with the Li-ion potential map calculated using BVSE (The yellow isosurfaces correspond to Li-ions probability density,indicating Li-ion conduction path).
尽管BVSE 方法能快速计算晶体中的Li 离子传输路径和能垒,但由于该理论忽略了结构弛豫,因此BVSE 方法计算的迁移能垒通常比第一性原理计算的值要大[50].在离子迁移的模拟方面,第一性原理计算被公认具有较高的精度,因此,我们利用基于DFT 的NEB 方法进一步计算了6 种组分的Li 离子迁移能垒.根据前面BVSE 的计算结果,可知LixYCl3+x(x=2.14,3.00,4.20)和LixYBr3+x(x=1.8,3.0,5.0)分别具有一维和三维扩散通道.为了节约计算资源,对LixYCl3+x(x=2.14,3.00,4.20)组分仅考虑了Li 离子在c方向的迁移路径及能垒,对LixYBr3+x(x=1.8,3.0,5.0)也只选择一条迁移路径进行研究.为了尽可能地避免其他因素的影响,我们对所有组分均选择了同一位置的Li 离子沿相同方向的迁移路径.例如,在体系LixYCl3+x(x=2.14,3.00,4.20)中,均选择同一位置的Li 离子沿c方向,以Oct (Liini)→Oct (Lifin)的路径迁移到相同的Li 空位,见图4 黑色虚线矩形框中的箭头.而在LixYBr3+x(x=1.8,3.0,5.0)中,选择的是在ab平面内、沿Oct (Liini)→Tet→Oct(Lifin)迁移的路径,如图5 红色虚线矩形框中的箭头所示.需要说明的是,由于Li15Y3Br24的Li 空位浓度比较低,在其基态结构中没有与Li9Y5Br24和Li12Y4Br24完全对应的迁移路径,因此,我们选择了同一位置的Li 离子 (Liini)在ab平面内向近邻Li 空位迁移的另一条路径,见图5(c).所有组分计算的迁移能垒如图9 所示.由图9(a)—(c)可知,Li15Y7Cl36,Li18Y6Cl36和Li21Y5Cl36的迁移能垒分别为0.26,0.19 和0.22 eV,结果表明Li18Y6Cl36配置展示出最低的迁移能垒.同样地,Li9Y5Br24,Li12Y4Br24和Li15Y3Br24的迁移能垒分别为0.38,0.24 和0.27 eV,Li12Y4Br24具有最低的迁移能垒.
根据前面的讨论,接下来将从离子间库仑相互作用的角度,对不同组分中迁移能垒差异的原因进行分析.首先,考虑了Li 离子在迁移过程中与配体阴离子的库仑作用.在LixYCl3+x(x=2.14,3.00,4.20)中,由于Li 离子沿Oct→Oct 的路径直接迁移,所以我们分别计算了Li15Y7Cl36,Li18Y6Cl36和Li21Y5Cl36三种组分中过渡态的Li 离子(Litr)与近邻三个Cl 离子的平均距离,其值分别为2.31,2.27,2.28 Å,如图9(a)—(c)中插图所示;对于LixYBr3+x(x=1.8,3.0,5.0),由于Li 离子迁移通过四面体间隙位,因此我们分别计算了Li9Y5Br24,Li12Y4Br24和Li15Y3Br24中过渡态Li 离子(Litr)形成的四面体的平均键长,分别为2.55,2.56 和2.52 Å,如图9(d)—(f)插图所示.我们发现,不管是氯化物还是溴化物,Li 离子在迁移过程与配体的相互作用变化都不大,这表明不同组分下配体对材料Li 离子迁移的影响应该不明显.进一步研究发现,Y3+离子占所有阳离子的比例,Li15Y7Cl36和Li9Y5Br24在同类材料中最高,分别为31.82%和35.71%,其他占比仅在16.67%—25%内.根据我们以前的研究结果,由于Y3+与Li+之间的库仑排斥作用,通道附近的Y3+对Li3YCl6和Li3YBr6中Li 离子的迁移都有阻塞作用[46].由此,我们推测Li15Y7Cl36和Li9Y5Br24的高迁移能垒应该是由于Y3+的浓度提高,进而显著增加了Y3+与Li+之间的库仑排斥力,导致加大了对Li 离子迁移的阻塞影响.如图4(a)和图5(a)所示,Y1原子位于Li 离子迁移通道附近,在这两个组分中,Y1-Lifin距离分别仅为3.64,3.84 Å,明显小于其他组分中的平均Y-Li距离,即3.78,4.05 Å.因此,Y1原子的占位将严重阻碍Li 离子的迁移.而对于Li9Y5Br24,这种阻塞效果将更明显.以Y1Br6八面体为例,该八面体附近的所有Li 离子四面体间隙通道,由于其与Y1的距离比较近,产生强的库仑排斥力,所以这些通道都将受到阻塞,这可能是单斜结构的溴化物比三角结构氯化物迁移能垒普遍高的一个主要因素.随着Y3+离子浓度的下降,即Li 离子浓度的增加,这种阻塞影响也随之降低,Li 离子迁移能垒从而显著降低.然而,Li 离子与Li 空位存在一个最佳平衡浓度,因为随着Li 离子浓度的不断提高,即Li 空位浓度不断降低,此时Li 离子可跳跃的空位数不断减少,伴随着可移动的Li 离子数量也将随之减少,故其能垒将会有所升高.当Li 离子与Li 空位浓度比值为3∶1 时,达到平衡,因此Li3YCl6以及Li3YBr6的迁移能垒最低.
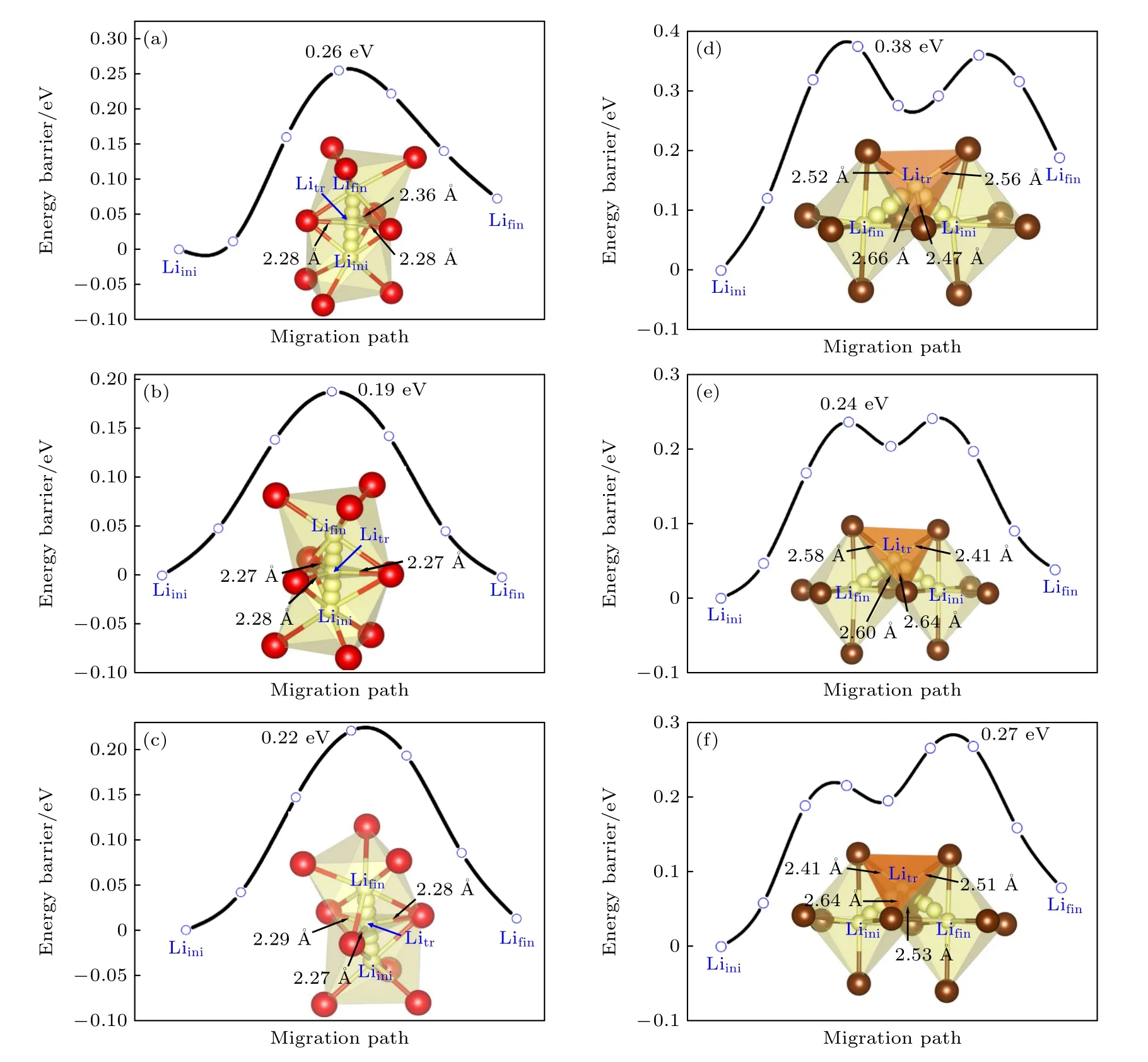
图9 所有组分的迁移能垒和局域结构示意图 (a) Li15Y7Cl36;(b) Li18Y6Cl36;(c) Li21Y5Cl36;(d) Li9Y5Br24;(e) Li12Y4Br24;(f) Li15Y3Br24Fig.9.Li-ion energy barrier profiles and local structures: (a) Li15Y7Cl36;(b) Li18Y6Cl36;(c) Li21Y5Cl36;(d) Li9Y5Br24;(e) Li12Y4Br24;(f) Li15Y3Br24.
主要采用第一性原理计算的方法研究了Li离子和Li 空位浓度对LixYCl3+x(x=2.14,3.00,4.20)和LixYBr3+x(x=1.8,3.0,5.0)材料结构、电子性质和离子迁移特性的影响.结果表明,所有组分均处于亚稳相,且都能较好地保持HCP 或CCP 阴离子排布;并且随着x的增大,材料的结构稳定性不断提高、带隙不断增大和迁移能垒不断下降.研究结果进一步表明,两类材料中Li 离子与Li 空位存在最佳平衡浓度.处于最佳平衡浓度下的Li3YCl6和Li3YBr6表现出较好的性能(图10).在同类材料中,Li3YCl6和Li3YBr6具有最大的带隙,即5.43,4.69 eV,表明其具有最大的理论电化学窗口;Li3YCl6和Li3YBr6的迁移能垒最低,分别为0.19,0.24 eV.此外,研究发现,Y3+浓度和占位对Li 离子的迁移影响较大,Y3+浓度越高,对Li 离子迁移的阻塞越大,迁移能垒越高.本文研究结果将为改善卤化物的性能及设计最佳组分的卤化物固体电解质提供一种新思路和新策略.
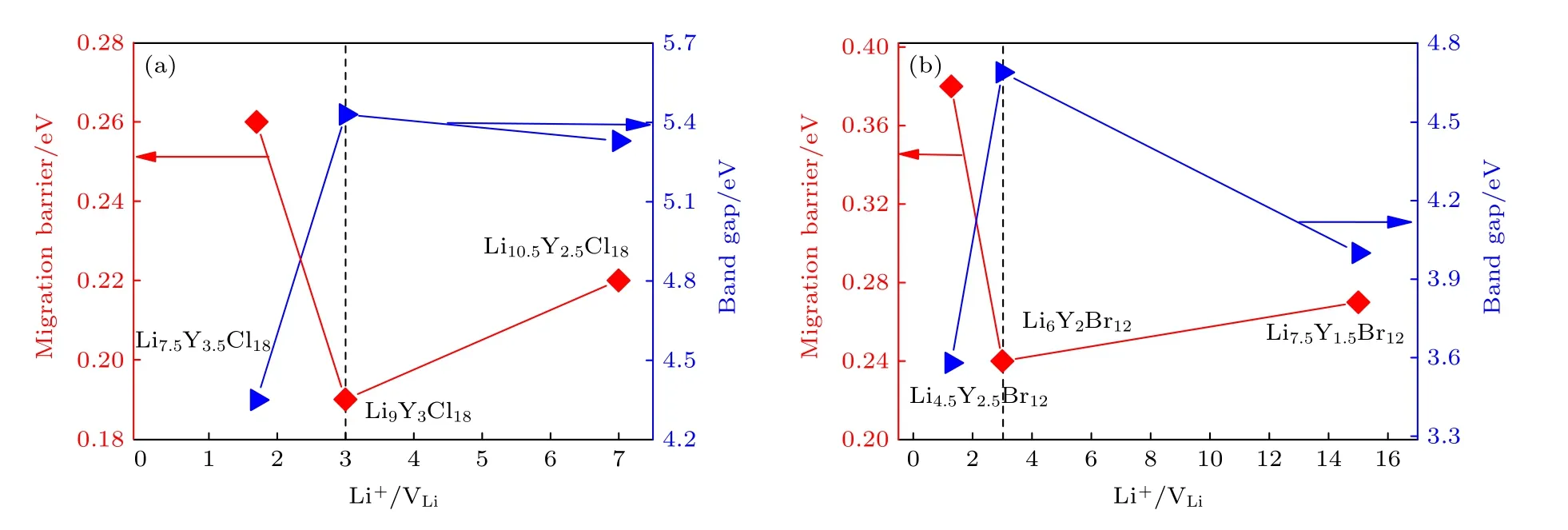
图10 (a) LixYCl3+x 和(b) LixYBr3+x 中迁移能垒和带隙随Li 离子与Li 空位(Li+/VLi)比例的变化关系Fig.10.Migration barriers and band gaps of (a) LixYCl3+x and (b) LixYBr3+x as a function of Li+/VLi ratio.
猜你喜欢 空位电导率组分 近红外定标法分析黏/锦/氨三组分纤维含量纺织标准与质量(2022年2期)2022-07-12容重及含水率对土壤电导率的影响研究干旱地区农业研究(2022年3期)2022-05-24稻米氨基酸含量和组分及其调控作物学报(2022年5期)2022-03-16煤的族组分基本特性研究当代化工(2019年3期)2019-12-12Analysis of Pragmatic Conditions of Null Subject in Mandarin校园英语·月末(2019年11期)2019-09-10不同低温处理对桃1年生枝相对电导率的影响山西果树(2017年4期)2018-02-082265FS土壤原位电导仪测定结果与土壤含盐量的关系湖北农业科学(2014年13期)2014-08-28空位读者欣赏(2014年6期)2014-07-03组分对PI/TiO2纳米复合薄膜电学性能的影响哈尔滨理工大学学报(2014年1期)2014-06-23时效制度对7075合金电导率的影响科技致富向导(2013年12期)2013-07-05